

Dose Matters: Navigating FDA Guidance for Oncology Drug Development – Part 2
Part 2 – Modelling a Better Dose: Using MIDD to meet Regulatory Expectations & Make Smarter Decisions in Drug Development
In the previous blog post, we introduced the FDA guidance “Optimizing the Dosage of Human Prescription Drugs and Biological Products for the Treatment of Oncologic Diseases”, concepts related to Project Optimus and how these impact different parts of the industry.
This time, we want to dig deeper into how Model Informed Drug Development (MIDD) can help trial sponsors adhere to the FDA recommendations and ensure dose optimisation is a central focus throughout oncology drug development.
The International Council for Harmonisation (ICH) defines MIDD as:
Practically, this means modelling allows you to create a ‘knowledge’ framework to understand the quantitative relationships of your drug and consider the totality of evidence in one system.
The essential benefits of applying MIDD in dose optimisation
-
- PK/PD modelling (with pre-clinical and clinical PK/PD data) helps to understand dose-exposure-response relationships and integrate biomarker-based measurements, where PD can refer to efficacy measures, clinical endpoints, or biomarker levels for safety or efficacy, etc.
- Key factors influencing these relationships, such as patient characteristics, cancer cell lines, co-medications and clinical biomarkers, can be identified and their impact assessed using population PK/PD modelling.
- Furthermore, MIDD allows you to visualise predicted outcomes for different design scenarios and offer a transparent and data-driven justification for efficient trial designs and dose range finding studies, an important practical and financial consideration.
- Altogether, this helps to guide dosage optimisation (which considers both dose and schedule) throughout drug development, in order to reduce unnecessarily high patient dosing and maximise patient benefit by maintaining efficacy while minimising safety risks.

MIDD approaches are relevant to all drug developers and should be applied across the full development life cycle to maximise benefit
As a key component of the guidance on dose optimisation and increasingly considered an expectation from regulatory bodies, MIDD approaches should now be considered by all drug developers, whether in large pharma or a newly established biotech.
Incorporating MIDD into your development plan brings advantages beyond just meeting regulatory expectations; it can generate the required evidence to support better-informed decisions that can reduce risk across the entire drug development paradigm.
Here we will look at how modelling can be used across the different stages of drug development to ultimately support the dose optimisation process:
-
- Pre-FIH Trial & Preparing for IND Submission
- Phase 1 & Selecting Recommended Phase 2 Dose (RP2D)
- Phase 2 & Setting your drug up for clinical success
- Pre-FIH Trial & Preparing for IND Submission
PK/PD modelling before FIH trials helps bridge preclinical data to the clinical setting, supporting robust dose selection for IND submission. It integrates preclinical PK data — by methods such as in vitro–in vivo extrapolation (IVIVE) and, where appropriate, allometric scaling — alongside pharmacodynamic and tolerability data. This includes evidence from receptor occupancy studies, efficacy studies, and biomarker responses. Together, these inputs allow modelling to translate preclinical exposure–response relationships into predictions for human response. This supports the selection of a safe and appropriate clinical starting dose, aligned with approaches such as the Minimal Anticipated Biological Effect Level (MABEL) and exposure matching.
Incorporating mechanistic or semi-mechanistic models to account for pharmacogenomic and population variability can further strengthen IND submissions, demonstrating a well-supported dose rationale.
Simulations of the model developed can be used to refine design of your Phase 1 study, including dose escalation strategies and safety monitoring plans, and can also be useful to address questions from regulatory agencies. Additionally, more complex drug-scheduling scenarios, that account for time dependencies, or treatment sequencing (see Case Study 1) can be evaluated in order to increase the likelihood of improved outcomes, e.g. survival rates.
Incorporating PK/PD modelling at this stage can help to de-risk your FIH study, maximising confidence in dosing strategies both with internal stakeholders and regulators and streamline your path to clinical development and IND approval.

2. Phase 1 & Selecting Recommended Phase 2 Dose (RP2D)
In Phase 1, PK/PD modelling is essential for transitioning from safety-focused dosing to selecting the Recommended Phase 2 Dose (RP2D) using robust, data-driven insights. The FDA guidance encourages evaluation of multiple dose levels, considering not just efficacy but also tolerability to establish comprehensive understanding of dose-exposure-response relationships. PK/PD modelling enables the integration of this early clinical data with preclinical findings to further explore and understand dose- and exposure-response relationships.
In addition to PK/PD modelling, additional quantitative methods to provide insights on inter-individual variance, i.e. how different patients (or subpopulations) might respond to a given dose, can also be developed at this stage. Population PK approaches should ideally be developed to give early insight and can be subsequently refined as increasing clinical data becomes available throughout development.
These methods can help to understand how intrinsic factors – such as body weight, age, sex, race and ethnicity, organ impairment, or genetic variation – as well as extrinsic factors, such as concomitant medications or food effects, influence drug pharmacokinetics. Where these factors result in clinically meaningful differences in exposure, population PK methods can support identification of subgroups that may require additional management strategies. Benefits are particularly strong when sampling data might be restricted, such as for highly toxic drugs or in a paediatric population.
Application of MIDD approaches at this stage may also provide critical input for designing expansion cohorts, exploring novel endpoints, understanding the impact of loading dose and evaluation of alternate dosing strategies such as intra-patient escalation or titration, and facilitating combination doses & schedules.
By leveraging these insights, sponsors can gain greater confidence in the Optimal Biological Dose (OBD), avoiding under- or over-dosing — both of which can risk trial failure. A clearly identified OBD enables selection of an evidence-based RP2D, allowing for more focused trial designs, recruiting fewer patients while maximising the likelihood of Phase 2 success.

- Phase 2 & Setting your drug up for clinical success
Phase 2 represents a critical juncture for refining dosing strategies and preparing for registrational trials. By integrating Phase 1 data with emerging clinical data, PK/PD models can be used to simulate “what if” scenarios such as dosing for specific patient populations, and predict tumour response, progression-free survival, and potential adverse effects to allow protocol refinement or dose modifications to be guided by quantitative approaches that can maximise clinical benefit. Adaptive trial designs informed by modelling can further refine dosing regimens, reducing the likelihood of trial failure in Phase 3.
Early insights into patient subgroups likely to benefit most can also guide the strategy for personalised medicine, supporting regulatory discussions and ensuring data-driven decision-making throughout development.
The insights provided by modelling at this stage informs the selection of optimal dosing for Phase 3 trials, improving trial success rates and reducing costly trial failures.

Balancing benefit and risk
Ideally, dose optimisation should allow for balancing benefit and risk. Developing aggregated metrics, or multi-objective optimization approaches (Lyons, 2021) that can be more readily interpretable can be implemented with methods such as Clinical Utility Indices (CUIs) (Ouellet, 2010).
Such methods can be useful for comparison of treatments, such as a recent study by Asiimwe et al, 2025. Here they assessed dosages to determine which provided the greatest benefit-risk profile for two ADC treatments, by assessing a CUI that provided a composite score of effectiveness (e.g. Overall Response Rate (ORR)) and toxicity (Dose Limiting Toxicities (DLTs)).
CUIs create a framework that allows multiple stakeholders to contribute to dose selection decisions, for example not only clinicians but also patients by considering efficacy and safety alongside quality of life.
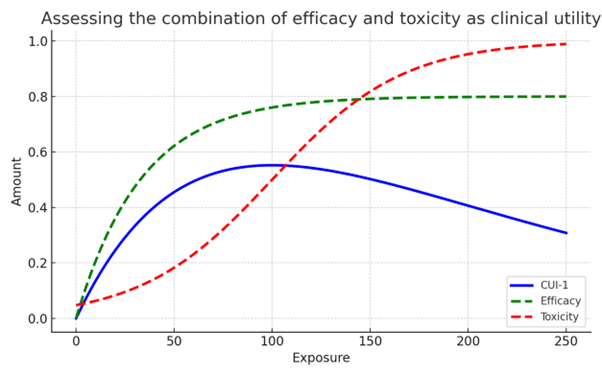
Figure 1. Plot shows an example of how combining 2 or more clinically meaningful parameters, such as efficacy and safety into a single metric (CUI) can be helpful for dosing decisions.
Conclusion: Embedding MIDD into your development strategy
MIDD provides a robust approach to dose optimisation across the entire development lifecycle. Applied early and iteratively, it enables integration of emerging data to guide dose and schedule decisions, helping developers reduce risk, meet regulatory expectations, and improve the likelihood of clinical success from early on.
While we and the FDA are encouraging early adoption of MIDD into drug development there are also applications in the post-marketing setting, such as in personalised dosing to improve individual outcomes. This is particularly important from a patient perspective in cancer care when historically drugs have entered the market often only providing marginal advantages (Villette et al, 2023).
Embedding MIDD into your development strategy is not just about regulatory alignment; it’s about making smarter, more patient-centred decisions that reduce risk, increase efficiency, and ultimately improve outcomes.
Furthermore, MIDD approaches can help facilitate early FDA discussions, demonstrating a thorough and proactive clinical development plan and a pragmatic approach to regulator interactions prior to availability of mature clinical data.

Looking to strengthen your dose optimisation strategy with robust, model-based evidence?
Whether you’re planning your first-in-human study, selecting your RP2D, or preparing for registrational trials, MIDD can help you make smarter decisions, reduce development risk, and engage effectively with regulators.
Reach out to Physiomics or Weatherden to explore how tailored quantitative approaches can support your programme and align with the latest FDA guidance.
This article was co-authored by team members at Physiomics plc and Weatherden: Hayley Close, Mark Davies, Michael Grant, and Simon Hutchings.
